Editorial
May 2021, 30:1
First online: 24 May 2021
Editorial
Clinical Implications of Genetic Profiling, Polygenic Risk Scores, LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease (ASCVD) – What is New?
David KL Quek, MBBS, FRCP, FNHAM, FAFPM (Hon.), FAsCC, FAPSC, FACC, FESC
LDL CHOLESTEROL (APOB) IS THE PREDOMINANT CAUSE OF ATHEROSCLEROSIS, ASCVD
Elevated levels of circulating Low-density lipoprotein (LDL) is now considered as the predominant putative cause of Atherosclerotic Cardiovascular Disease (ASCVD)1 We now recognise that LDL-cholesterol is central to the atherosclerotic process. The Lipid-Atherosclerosis hypothesis has been well established. Yet there is confusion and controversy as to its causative role with many physicians still implying that inflammatory processes may play a bigger role.2
Of course, immunity and inflammation further contribute to the acceleration of atherogenesis, but almost invariably, this is in the context of an underlying dyslipidemic milieu.3 The reality is that LDL-cholesterol in high circulating levels is the main driver and trigger for almost all biological and human ASCVD. Both innate and adaptive immune responses are triggered that lead to upregulation of cell-adhesion molecules (iCAM, P-selectin and CVAM-1), T-helper cell-, T-regulatory cell- and B lymphocyte mediated cytokines, which in turn attract circulating monocytes to adhere to and phagocytose oxidized LDL particles, leading ultimately to the formation of foam cells and further igniting inflammatory responses that damage the endothelium.4, 5
LDL-cholesterol is the predominant (90-96%) underlying circulating molecule for apolipoprotein-B, which causes atherogenesis and subsequently ASCVD.6 Apolipoprotein-B-containing LDL (ApoB-LDL) particles infiltrate and get retained in the vascular endothelium. This ApoB-containing LDL retention in the subintima results in loss of endothelial integrity and function. The LDL-ApoB particles trigger atherosclerotic plaque formation, by stimulating the cascade of immune and inflammatory processes.
LDL-cholesterol and its link with ASCVD indeed passes all the eight Hill criteria7 for causality in disease association. 1) LDL is the plausible putative pathologic mechanism that leads to the atherosclerotic process;8 2) LDL has a strong graded relationship to the atherosclerotic process and disease;9, 10, 11, 12, 13, 14, 15, 16 3) there is a magnitude/duration-dependent LDL-level biological gradient: i.e., graded risk factor elevation that precedes disease;7, 8, 17 4) LDL to atherosclerotic disease has a direct temporal effect sequence: independent from other confounding factors;7, 8-13 5) LDL-ASCVD association has shown robust consistency: there is consistent relationship across many strong RCT studies;7, 8-11, 18, 19 6) LDL linkage to ASCVD has shown coherence between different approaches; and 7) LDL-ASCVD linkage is increasingly supported and buttressed by very specific genetic findings.20, 21, 22 Finally, 8) there is graded reduction of ASCVD risk with intervention by lowering LDL: i.e., the reduction in circulating LDL is uniformly shown to decrease the incidence of ASCVD; this has now been rigorously evidenced by at least 28 huge of LDL-cholesterol lowering studies.13-16, 23, 24, 25, 26, 27
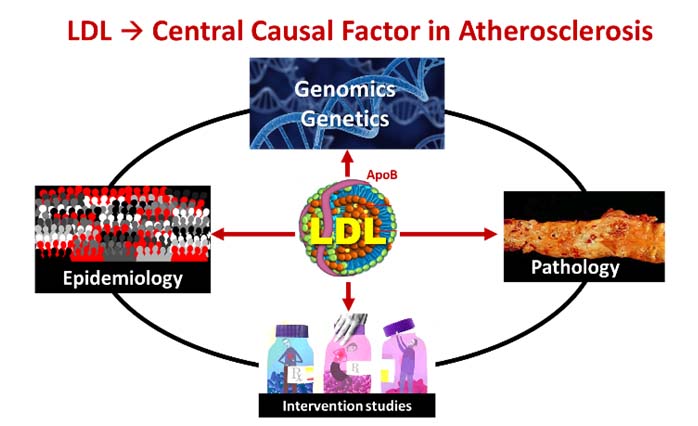

Figure 1
GENES AND THE LDL-MODEL OF ATHEROSCLEROSIS
The LDL-gene nexus began with the identification of the LDL-receptor by Goldstein and Brown.8 We have now identified several single nucleotide polymorphisms (SNPs) that are associated with variable phenotypic expressions of blood LDL levels and their links to coronary heart disease risk. Some of the more recogniseable SNPs include those related to SORT1, PCSK9, LDLR, HMGCR, ABCG8, APOE allele exposures.
Depending on the gene variations, gain-of-function (GOF) and loss-of-function (LOF) gene alleles give rise to a composite array of either very high or very low LDL phenotypic expressions, that are now consistently correlated with disease outcomes. These genetic evidence bases for the LDL paradigm linkage to ASCVD risk are compelling and intriguing.
One more notable cause celebre of gene-ASCVD linkage is that for familial hypercholesterolemia (FH). The commonest version of FH is a mutation of the LDLR gene, which leads to decreased or abnormal LDL-receptor function resulting in markedly increased blood concentration of circulating LDL particles. In the homozygous expression (HoFH), this abnormally high circulating LDL cholesterol (usually > 13 mmol/L from birth) has been famously associated with universal early development of ASCVD in childhood or adolescence. More common is the heterozygous FH (HeFH) that affects ≈1 in 250 people worldwide.28 Phenotypic expression of FH is variable and there is now putative proof that atherosclerosis and risk for cardiovascular events, is proportional both to the duration and magnitude of exposure to the high LDL-C environment.20, 29, 30
Ference et al16 have compellingly shown that genetic mutations of exposure alleles that result in various degrees of lower or higher LDL levels, correspond and correlate very closely with the actual degree of incident risk for coronary heart disease, initially using Genome-wide association study (GWAS) tools and SNPs.
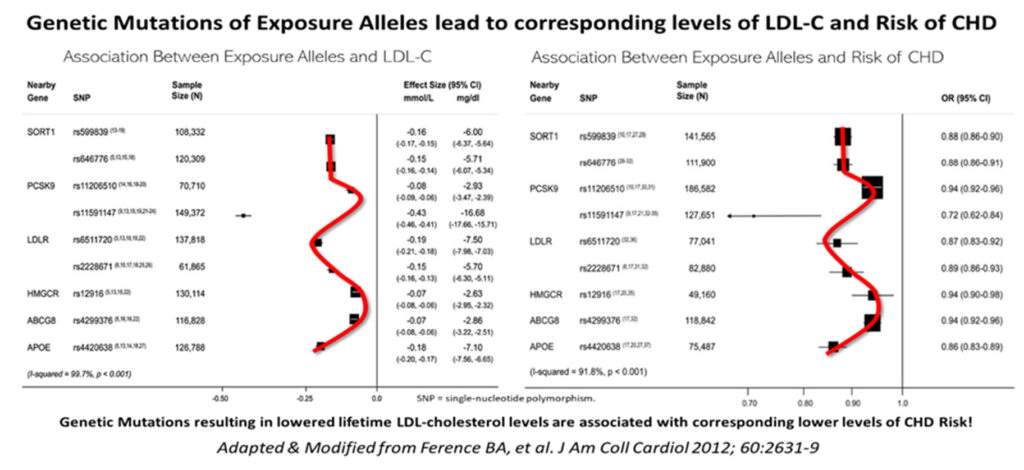
Another classic example of gene variation i.e., one related to Loss of Function (LOF), is that of the PSCK9 LOF (nonsense) mutation, rs11591147, which results in very low LDL levels; this in turn, is associated with very low lifetime risk and incidence of ASCVD.20
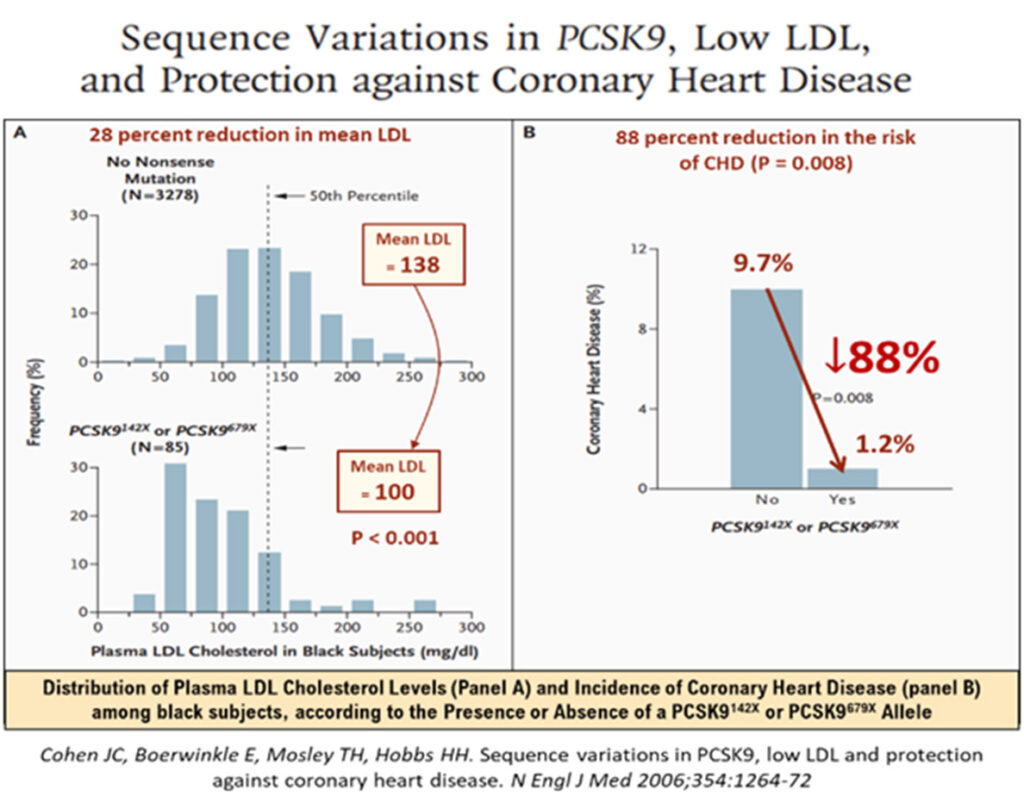
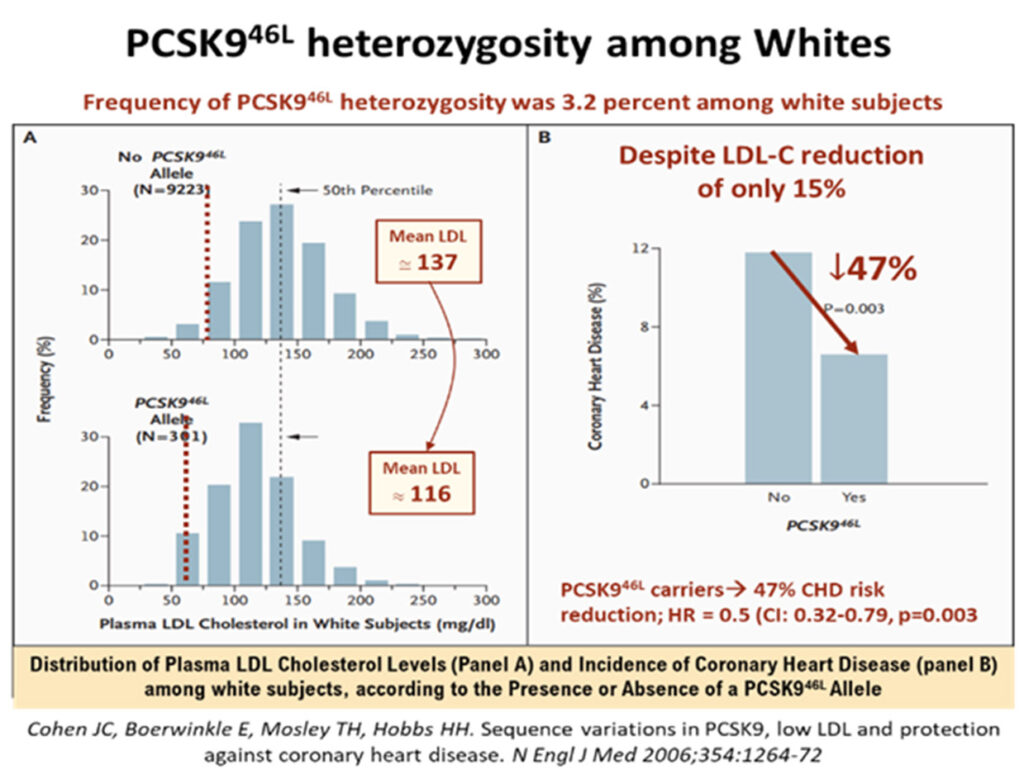
A 2006 study of rare PSCK9 allele mutations in some 2.6% of African Americans has shown that where there were nonsense mutation (PCSK9142X or PCSK9679X alleles), the mean LDL-cholesterol levels were low, down by about 28%, but were associated with very low incidence of CHD, a reduction by as much as 88%!31 Among white Americans, the prevalence of PCSK9 variations was also impressive, some 3.2% had the PCSK946L allele, that led to a mean LDL reduction (137 to 116mmg/dL) of ≈15%, but which was associated with robust CHD risk reduction of -47% (p = 0.003). The mean LDL cholesterol reductions were modest, but these phenotypic expressions of outcomes in real lives, underscore the fact that longer term exposure to these lower levels of LDL, were associated with lower attenuated lifetime risk of CHD.
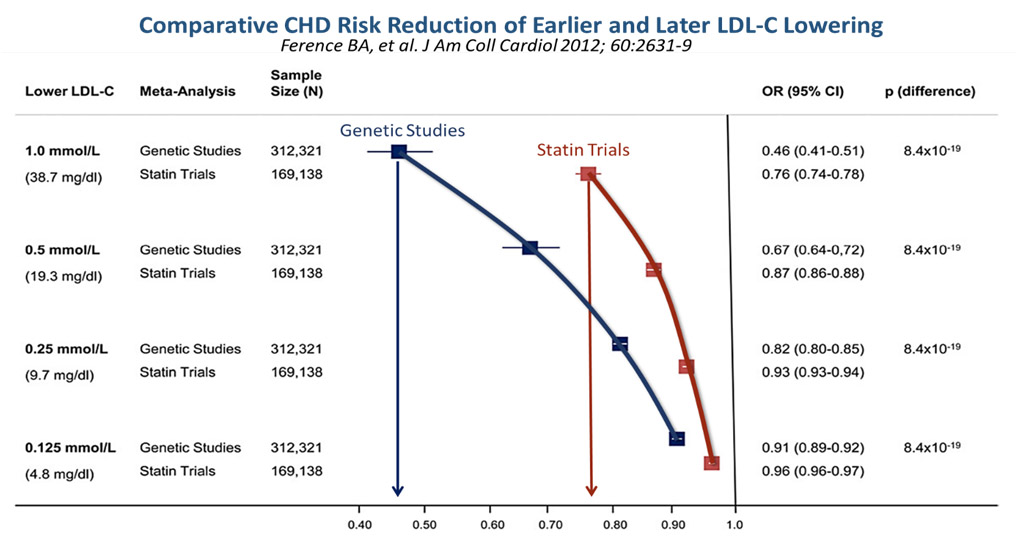
Thus, in their paper, Ference et al,16 has constructed a graphically attractive and intuitive concept that genetic analyses in people with heritable lower LDL levels always outperform the beneficial outcomes of LDL-cholesterol reduction with medications as shown in the statin trials. For example, for every 1.0 mmol/l reduction of LDL-C from genetic studies, there was a strongly significant Odds Ratio Reduction in risk for CHD of 0.46, versus that achieved by statin trials, which showed just 0.76, as seen in the Figure 4 below. Therefore, the earlier the LDL-C is reduced, (i.e., the longer lifetime exposure to lower LDL-C environment seen in heritable low LDL gene polymorphisms), the lower the risk for CHD or ASCVD!
Recently there have been several studies utilising genetic probes and gene-based profiling to determine their usefulness in improving our prediction of the highest ASCVD-risk individuals. Arguably, these would be the at risk population who would benefit most from more targeted and aggressive interventions.32, 33, 34, 35 So, is there any added value in having more refined risk profiling tools to help our risk stratification of the patients most in need of targeted therapies. Are genetic tools helpful?
POLYGENIC RISK SCORES & PROFILES
As it stands, there is a pervasive global disinformation campaign against the lowering of LDL cholesterol with medications, particularly with statins. Therefore, there is a need to counter these unfounded but viral social media hypes that restrain patients from adopting these life-modifying therapies more readily and with less qualms as to their supposed adverse side effects. Physicians are thus, urged to be more proactive in managing our higher risk patients more optimally. We do need more robust and convincing data and proof to help our patients choose the better options. We need to show that LDL-reduction therapy is solidly scientific and backed by some massive incontrovertible body of evidence.
Can the expanding interests in improving accuracy of risk prediction algorithms help in this endeavour? Should more and more polygenic risk scores be used to help us decipher who would benefit from more guideline-directed therapies? How do we translate this emerging science into clinical practice?36
This approach is somewhat controversial. While there has been much recent research to refine and improve the accuracy of predicting ASCVD risk, the benefits of various polygenic risk scores have not been very compelling. At best there is modest additive predictive value, but the ultimate benefits from such adoption may not be sufficiently cost effective or realistic. The question has always been how much do we as physicians need to do: how far do we need to go, to define, to test, to enact our diagnostic and therapeutic arsenals?
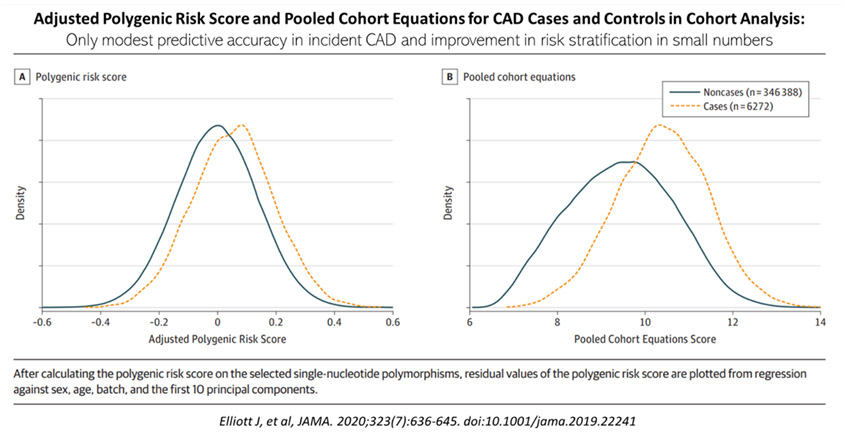
Elliot et al,30 performed a recent analysis of case-control sample of 15,947 prevalent CAD cases matched with a cohort of controls for polygenic risk score for CAD based on summary statistics from published genome-wide association studies (GWAS). A separate cohort of 352,660 individuals (sourced from UK Biobank participants enrolled from 2006 – 2010) was used to evaluate the predictive accuracy of the polygenic risk score (PRS), pooled cohort equations and both combined for incident CAD.
In the 352,660 cohort, there were 6272 incident CAD events over a median of 8-year follow-up. The C-statistics for the PRS, pooled cohort equations and both combined were 0.61, 0.76 and 0.78, respectively. Essentially, adding the PRS to the pooled cohort, resulted in a net reclassification improvement of 4.4% for cases and -0.4% for non cases, a net reclassification improvement of 4%, see Figure 5. Thus, there is only a very modest improvement in risk classification for ASCVD when PRS is added to the usual conventional risk factors of smoking, diabetes, hypertension, body mass index, family history of IHD, high blood cholesterol.
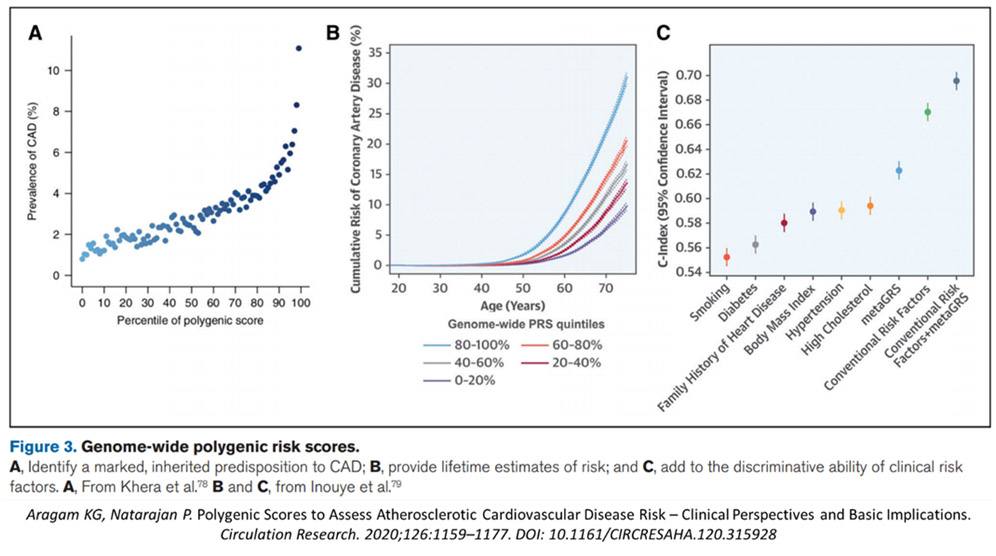
A more recent 2021 systematic review by Aragam and Natarajan,29 reinforced this impression. Here, they showed that there was modest improvement in predictive value adding the PRS to combined conventional risk factors, from a C-statistic of 067 to 0.69, slightly lower in this pooled data analysis (see Figure 6). Elliot et al25 concluded that “the use of genetic information over pooled cohort equations model warrants further investigation before clinical implementation”.
TRANSLATIONAL SCIENCE TO CLINICAL PRACTICE
How about secondary prevention? Can genetic probes help in making our therapeutic management pathways more evidence based? Can we improve our risk predictive accuracy particularly when we are faced with very high-risk patients with ASCVD, i.e., patients with life-threatening recurrent cardiovascular events, despite the usual optimal medical therapies?
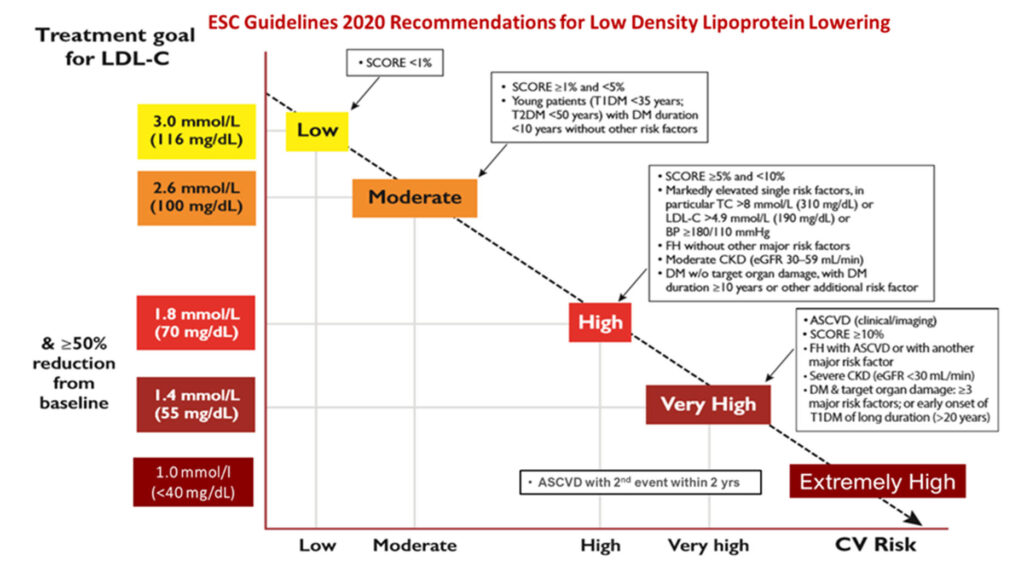
How far do we go in trying to reduce LDL cholesterol to the goals set by most cardiology guidelines, i.e., to below 1.4 mmol/l, or even <1.0 mmol/l, especially among those with back-to-back relapsing acute coronary syndromes within 2 years? Is there proof that lower is indeed better? Here, there is some evidence from the ODYSSEY Outcomes study.
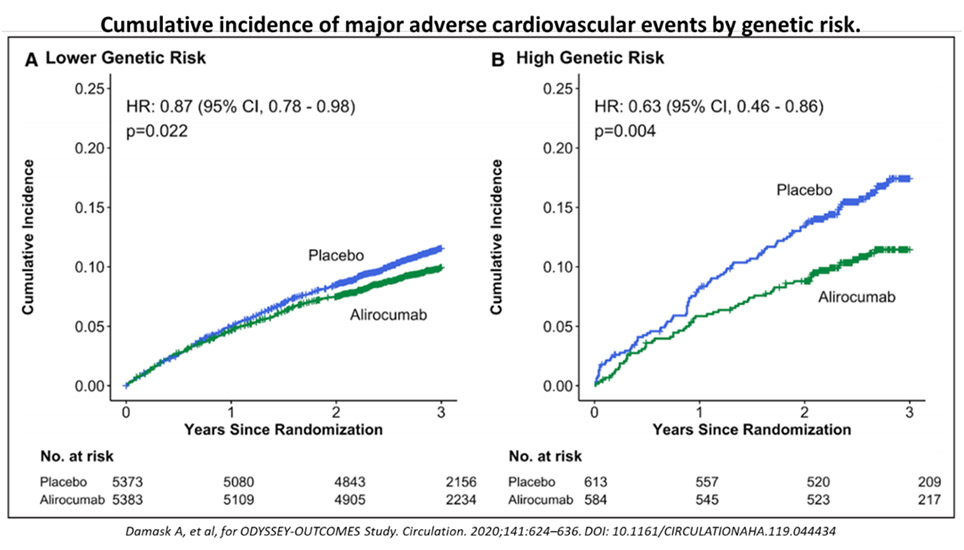
Damask, et al,37 recently analysed how using the genome-wide PRS for CAD may help patients receive greater clinical benefit from PCSK9-inhibitor treatment. The concept here is to determine if genetic approaches can help identify high risk patients as well as those who might benefit more from more aggressive or special therapies.38, 39 In this impressive study, there was an absolute reduction by alirocumab in high versus low PRS groups of 6.0% and 1.5% respectively, and a relative risk reduction by 37% vs. 13%, respectively (p = 0.04), see Figure 7. What this implies is that for very high-risk ASCVD patients, using the PRS can help us to identify those who would benefit more through more targeted therapies, and more aggressive lowering of LDL. Importantly, there is robust end-point MACE outcome benefit to show as well.
In the monoclonal antibody PCSK9-inhibitor studies,40, 41, 42, 43, 44 low levels of LDL <1.0 mmol/l (38 mg/dL) mg/dL) were also seen in some individuals, but these were not shown any offsetting safety concerns. If anything, their ASCVD risk was further reduced. In a recent review the safety of very low levels of LDL, Karagiannis et al,45 concluded that “given the potential for cardiovascular benefit and short-term safety profile of very low (≤30 mg/dL) LDL-C levels, it may be advantageous to attain such low levels in specific high-risk subsets of patients. Thus, polygenic risk profiling may help us determine these subsets of patients who would require the best benefits. Hence, the epigram that the lower the LDL, the better the ASCVD outcome!
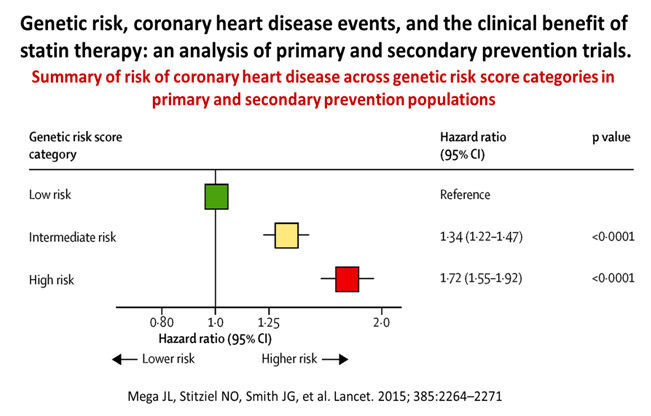
Thus, the lower the LDL, the better the ASCVD outcomes. This is not new, as earlier studies have also shown that patients with high PRS for CAD had higher relative and absolute risk reduction in cardiovascular events after statin treatment in both primary and secondary prevention settings. The meta-analysis and systematic review by Mega et al,46 combined and studied a community-based cohort study (the Malmo Diet and Cancer Study) and 4 trials, 2 primary prevention studies (JUPITER and ASCOT) and 2 secondary prevention studies (CARE and PROVE-IT-TIMI 22).
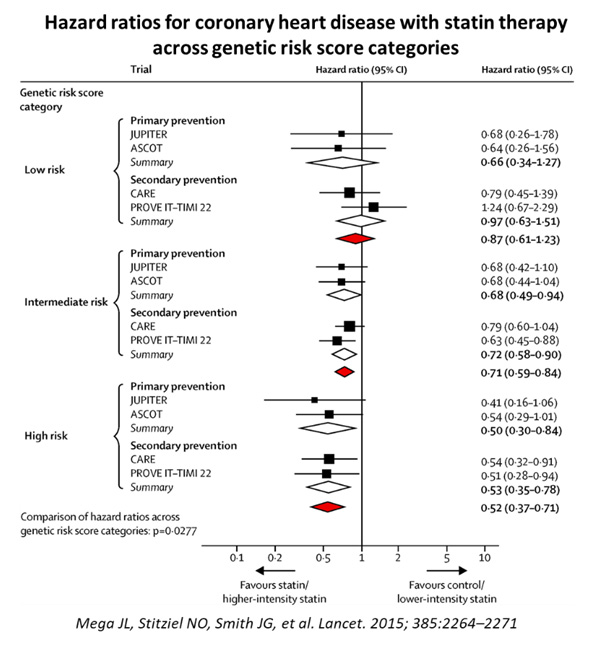
48,421 people were studied, and 3477 events were analysed, with the association of genetic risk score based on 27 genetic variants, adjusted for traditional clinical risk factors. Those with high PRS had a stepwise incremental multi-variable adjusted hazard ratio risk for incident CAD from a reference of 1.0 to 1.72 (See Figure 7). More impressive was the finding that statin use resulted in greatest relative risk reductions among those with highest genetic risk categories, from 48% to 29% and 13% for high, intermediate, and low risk categories, respectively (see Figure 8).
Perhaps as important, apart from clinical outcomes, is whether patients with high-risk atherosclerotic pathologies are improved, or can be shown to benefit from using these more elaborate PRS scores on top of conventional risk factors.
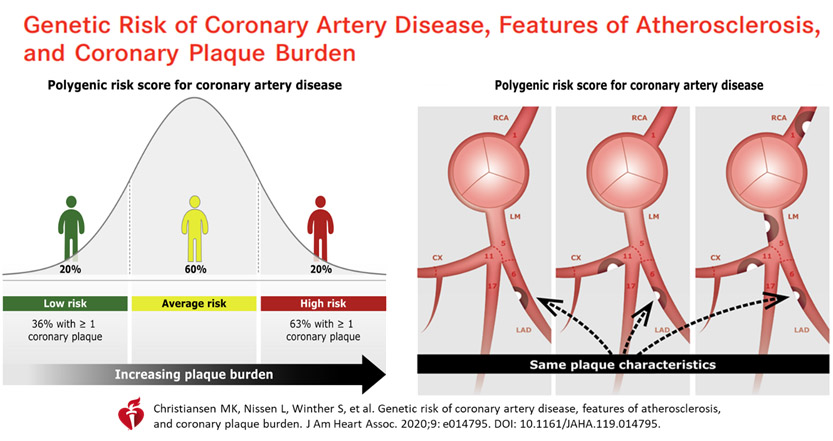
A recent Danish study47 explored PRS added on to conventional risk factors, to see if these were better at predicting the coronary plaque burden as assessed by coronary CT-angiography, i.e., improving plaque characteristics with more aggressive PRS-directed LDL lowering therapies. However, this was not shown. Although overall there was added PRS impact on better prediction of higher CAD risk, i.e., overall, more coronary atherosclerosis, there was no consistency in the segment-level analyses for plaque burden, severity, plaque composition and localization. Using CT angiography, it was not possible to show individual plaque pathology improvement, nor recognise at risk vulnerable plaques, even when guided by polygenic risk scores.
Therefore, PRS add-on impact for ASCVD Risk Stratification remains at best preliminary and intriguing in certain subsets of very high risk patients, who might otherwise receive less than optimum, less aggressive LDL lowering therapies.
DISCUSSION & CONCLUSION
Genetic profiling and the use of PRS has some modest effect on improving our predictive accuracy on high-risk individuals with regards incident ASCVD. However, overall, the clinical benefits have not been shown to be very compelling. For difficult patients with extremely high risk or recurrent cardiovascular sequelae or complications, the use of PRS added on to more traditional risk factor profiling can help convince patients, and physicians and third-party healthcare payers, as to the benefits of more aggressive lowering of the LDL cholesterol.
Aggressive treat-to-target statin, with possible combinations of ezetimibe, and PCSK9-inhibitor regimens to lower LDL cholesterol, have been shown to better guided by use of polygenic risk score profiling on top of clinical conventional risk factors. This is the basis for current guidelines on how much to reduce the LDL cholesterol levels, and to which ‘ideal’ targeted goals for different patient risk characteristics/profiles. Ultimately, extremely high-risk patients for ASCVD need the most evidence based risk stratification and therapies that we can offer.
While genetic profiling can help to bolster our efforts to initiate and be more aggressive in our efforts to reduce the LDL and ApoB in our higher risk patients, they remain as esoteric tools for a smaller number of individuals who need more convincing to lower their numbers well below the comfort levels for most physicians. For this group of individuals, they may require better risk stratification. Nevertheless, currently, we do have sufficient data to advocate for stronger collective efforts in helping to ameliorate or eradicate the mounting ASCVD crisis, globally.
The sensible implication is for us as clinicians to begin a conversation with our now increasingly statin-sceptical patients. In a recent update on the consensus guidelines regarding ASCVD and the role of LDL-ApoB, the EAS panel once again reiterate the causality of LDL in ASCVD: “extensive evidence from epidemiologic, genetic, and clinical intervention studies has indisputably shown that low-density lipoprotein (LDL) is causal in this process.”
We need to initiate LDL-C reduction therapies earlier rather than wait and try out other less effective measures, as so often demanded by our uninformed patient. We need to convince them about the futility of pandering to some unproven natural or alternative methods that clearly do not work as well, or are plain ineffective!
It is time to step up our efforts to reduce the scourge of ASCVD globally as well as in our own backyard. It is not uncommon to see many aspiring interventional cardiologists who prescribe the minimal statin dose post-ACS or post-PCI, rather than the guideline-directed correct dose and approach. The latest ESC guidelines remain a robust working paper with which to guide the optimum approach to best manage our patients, at the current status of our knowledge base.
A recent review by Robinson et al,48 on the overwhelming evidence for lipid lowering strategies, once again repeated the call to start early to help eradicate the burden of ASCD by lowering ApoB-containing lipoproteins earlier in life! Robinson and Gidding49 had first proposed in 2014 that curing atherosclerosis should be the next major cardiovascular prevention goal. In 2021, together with multinational group of clinician-scientists, they now reiterate their call that we must start translating the evidence into the next prevention paradigm i.e., ASCVD eradication. To achieve this, we must start to lower elevated LDL-C from a younger age, so that we can reduce the clinical manifestations of life threatening and debilitating atherosclerotic cardiovascular disease. The longer the duration and the greater the magnitude of exposure to an environment of elevated LDL cholesterol levels, the greater the ASCVD risk. So, we do need to start LDL-lowering intervention earlier!
We need to convince ourselves to do what is necessary, initiate statins or more, to help achieve appropriate guideline-targeted goals. The multitude of evidence is incontrovertible. With the current deluge of so much robust genetic evidence and polygenic risk probes, we have further strengthened and refined our understanding of this modern-day scourge even more. LDL-C reduction as one of the most effective strategies to lower ASCVD risk, has stood the test of time amidst massive evidence bases! And we need to start the treatment early and optimally!
REFERENCES
1. Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459-72. Doi:10.1093/eurheartj/ehx144 CrossRef Pubmed
2. Libby P. Inflammation in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045-2051. CrossRef Pubmed
3. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis Circ Res. 2019;124:315-327. CrossRef Pubmed
4. Hansson GK, Libby P. The immune response in atherosclerosis: a doubleedged sword. Nat Rev Immunol 2006;6:508–19. CrossRef Pubmed
5. Libby P, Ridker PM, Hansson GK. Inflammation in Atherosclerosis – From Pathophysiology to Practice. J Am Coll Cardiol 2009;54:2129–38. CrossRef Pubmed
6. Sniderman AD, Thanassoulis G, Glavinovic T, Navar AM, Pencina M, Catapano A, Ference BA. Apolipoprotein B Particles and Cardiovascular Disease. A Narrative Review. JAMA Cardiol. doi:10.1001/jamacardio.2019.3780. Published online October 23, 2019. CrossRef Pubmed
7. Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295-300
8. Goldstein JL , Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell 2015;161:161–172. CrossRef Pubmed
9. Raal FJ , Pilcher GJ, Waisberg R, Buthelezi EP, Veller MG, Joffe BI. Low-density lipoprotein cholesterol bulk is the pivotal determinant of atherosclerosis in familial hypercholesterolemia. Am J Cardiol 1999;83:1330–1333. CrossRef Pubmed
10. Schmidt HH , Hill S, Makariou EV, Feuerstein IM, Dugi KA, Hoeg JM. Relationship of cholesterol-year score to severity of calcific atherosclerosis and tissue deposition in homozygous familial hypercholesterolemia. Am J Cardiol 1996;77:575–580. CrossRef Pubmed
11. CARDIoGRAMplusC4D Consortium. A comprehensive 1000 Genomesbased genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130. CrossRef Pubmed
12. Prospective Studies Collaboration, Lewington S, Whitlock G, Clarke R, Sherliker P, Emberson J, Halsey J, Qizilbash N, Peto R, Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet 2007;370:1829–1839. CrossRef Pubmed
13. Lawlor DA , Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 2008;27:1133–1163. CrossRef Pubmed
14. Benn M , Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J 2016;37:1384–1394. CrossRef Pubmed
15. Emerging Risk Factors C , Di Angelantonio E, Gao P, Pennells L, Kaptoge S, et al. Lipid-related markers and cardiovascular disease prediction. JAMA 2012;307:2499–2506. CrossRef Pubmed
16. Ference BA , Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol 2012;60:2631–2639. CrossRef Pubmed
17. Cannon CP , Blazing MA, Giugliano RP, et al. IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015;372:2387–2397. CrossRef Pubmed
18. Sabatine MS, Giugliano RP, Keech AC, et al, FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017; doi: 10.1056/NEJMoa1615664. CrossRef Pubmed
19. Nicholls SJ , Puri R, Anderson T, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients. The GLAGOV randomized clinical trial. JAMA 2016;316:2373–2384. CrossRef Pubmed
20. Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, Voros S, Giugliano RP, Davey Smith G, Fazio S, Sabatine MS. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 2016;375:2144–2153. CrossRef Pubmed
21. Holmes MV, Asselbergs FW, Palmer TM, et al, for UCLEB Consortium. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J 2015;36:539–550. CrossRef Pubmed
22. Khera AV, Won HH, Peloso GM, et al. Diagnostic yield of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol 2016;67:2578–2589. CrossRef Pubmed
23. Lipid Research Clinics Program. The Lipid Research Clinics coronary primary prevention trial results: reduction in the incidence of coronary artery disease. JAMA 1984;251:351–364. CrossRef Pubmed
24. Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet 2010;376:1670–1681. CrossRef Pubmed
25. Baigent C, Landray MJ, Reith C,,et al., for the SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011;377:2181–2192. CrossRef Pubmed
26. Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med 2011;365:2078–2087. CrossRef Pubmed
27. Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, Sabatine MS. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA 2016;316:1289–1297. CrossRef Pubmed
28. Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science. 2016; 354:aaf7000. doi:10.1126/science.aaf700 CrossRef Pubmed
29. Raal FJ, Pilcher GJ, Waisberg R, Buthelezi EP, Veller MG, Joffe BI. Low-density lipoprotein cholesterol bulk is the pivotal determinant of atherosclerosis in familial hypercholesterolemia. Am J Cardiol 1999;83:1330–1333. CrossRef Pubmed
30. Schmidt HH, Hill S, Makariou EV, Feuerstein IM, Dugi KA, Hoeg JM. Relationship of cholesterol-year score to severity of calcific atherosclerosis and tissue deposition in homozygous familial hypercholesterolemia. Am J Cardiol 1996;77:575–580. CrossRef Pubmed
31. Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL and protection against coronary heart disease. N Engl J Med 2006;354:1264-72. CrossRef Pubmed
32. Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–1257. doi:10.1093/cvr/cvy084 CrossRef Pubmed
33. Aragam KG, Natarajan P. Polygenic Scores to Assess Atherosclerotic Cardiovascular Disease Risk – Clinical Perspectives and Basic Implications. Circulation Research. 2020;126:1159–1177. DOI:10.1161/CIRCRESAHA.120.315928 CrossRef Pubmed
34. Elliott J, Bodinier B, Bond TA, et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA. 2020;323(7):636-645. doi:10.1001/jama.2019.22241 CrossRef Pubmed
35. Bolli A, Domenico PD, Pastorino R, et al. Risk of Coronary Artery Disease conferred by Low-Density Lipoprotein Cholesterol depends on polygenic background. Circulation. 2021;143:1452–1454. DOI:10.1161/CIRCULATIONAHA.120.051843 CrossRef Pubmed
36. Knowles JW, Ashley EA. Cardiovascular disease: the rise of the genetic risk score. PLoS Med. 2018; 15:e1002546. doi:10.1371/journal. pmed.1002546 CrossRef Pubmed
37. Damask A, Steg PG, Schwartz GG, et al, for ODYSSEY-OUTCOMES Study. Patients with high Genome-Wide Polygenic Risk Scores for Coronary Artery Disease may receive greater clinical benefit from alirocumab treatment in the ODYSSY-OUTCOMES Trial. Circulation. 2020;141:624–636. DOI: 10.1161/CIRCULATIONAHA.119.044434 CrossRef Pubmed
38. Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19:581–590. doi:10.1038/s41576-018-0018-x CrossRef Pubmed
39. Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22. doi:10.1016/j.ajhg.2017.06.005 CrossRef Pubmed
40. Ginsberg, HN, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia and LDL-C of 160 mg/dl or higher. Cardiovasc. Drugs Ther. 2016;30,473–483. CrossRef Pubmed
41. Sabatine MS, Giugliano RP, Keech AC, et al, for the FOURIER Steering Committee and Investigators. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med 2017; 376:1713-1722. CrossRef Pubmed
42. Giugliano RP, Keech A, Murphy SA, et al. Clinical efficacy and safety of evolocumab in high-risk patients receiving a statin: secondary analysis of patients with low LDL cholesterol levels and in those already receiving a maximal-potency statin in a randomized clinical trial. JAMA Cardiol. 2017; 2,1385–1391. CrossRef Pubmed
43. Ference BA, Cannon CP, Landmesser U, Luscher TF, Catapano AL, Ray KK. Reduction of low density lipoprotein-cholesterol and cardiovascular events with proprotein convertase subtilisin-kexin type 9 (PCSK9) inhibitors and statins: an analysis of FOURIER, SPIRE, and the Cholesterol Treatment Trialists Collaboration. Eur. Heart J. 2018;39,2540–2545. CrossRef Pubmed
44. Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol 2019;16,155–165. https://doi.org/10.1038/s41569-018-0107-8 CrossRef Pubmed
45. Karagiannis AD, Mehta A, Dhindsa DS, et al. How low is safe? The frontier of very low (<30 mg/dL) LDL cholesterol. European Heart Journal (2021) 00,1–16. doi:10.1093/eurheartj/ehaa1080 CrossRef Pubmed
46. Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015; 385:2264–2271. doi:10.1016/S0140-6736(14)61730-X CrossRef Pubmed
47. Christiansen MK, Nissen L, Winther S, et al. Genetic risk of coronary artery disease, features of atherosclerosis, and coronary plaque burden. J Am Heart Assoc. 2020;9:e014795. DOI:10.1161/JAHA.119.014795. CrossRef Pubmed
48. Robinson JG, Williams KJ, Gidding S, et al. Eradicating the Burden of Atherosclerotic Cardiovascular Disease by Lowering Apolipoprotein B Lipoproteins Earlier in Life. J Am Heart Assoc. 2018;7:e009778. DOI: 10.1161/JAHA.118.009778 CrossRef Pubmed
49. Robinson JG, Gidding SS. Curing atherosclerosis should be the next major cardiovascular prevention goal. J Am Coll Cardiol. 2014;63:2779–2785. CrossRef Pubmed
Copyright Information